Just another WordPress site - Ruhr-Universität Bochum
Research Group
Atomistic Simulation of Thermodynamic Properties
 RUB, Marquard
RUB, MarquardResearch Group Leader
Room: IC 02-719
Tel.: +49 234 32 22572
E-Mail: sarath.menon@rub.de
Research
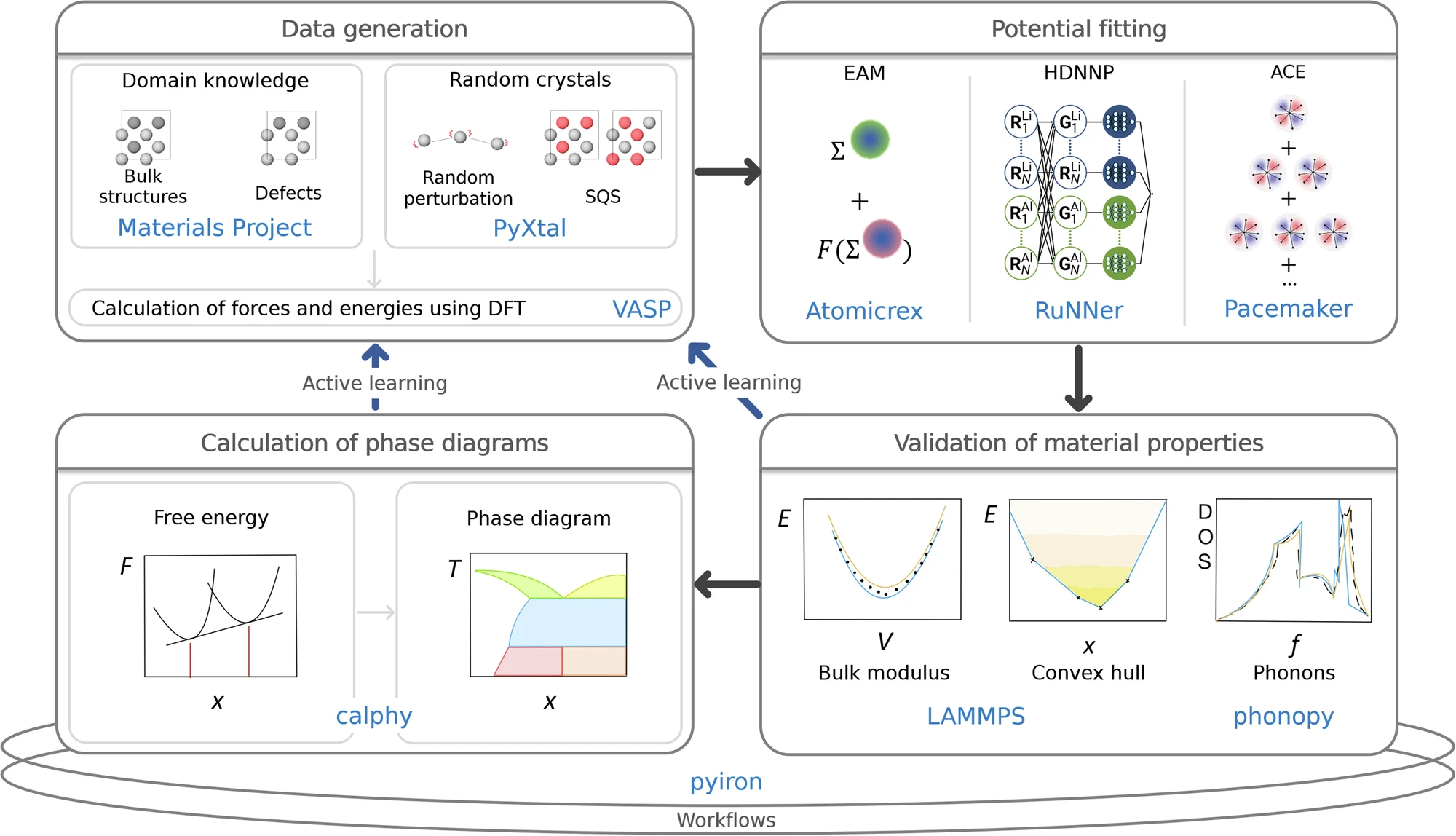
Calculation of thermodynamic properties and phase diagrams from atomistic simulations is essential to materials discovery and design, because it enables prediction of formation energies, phase stability and free energy landscapes directly from atomic interactions.
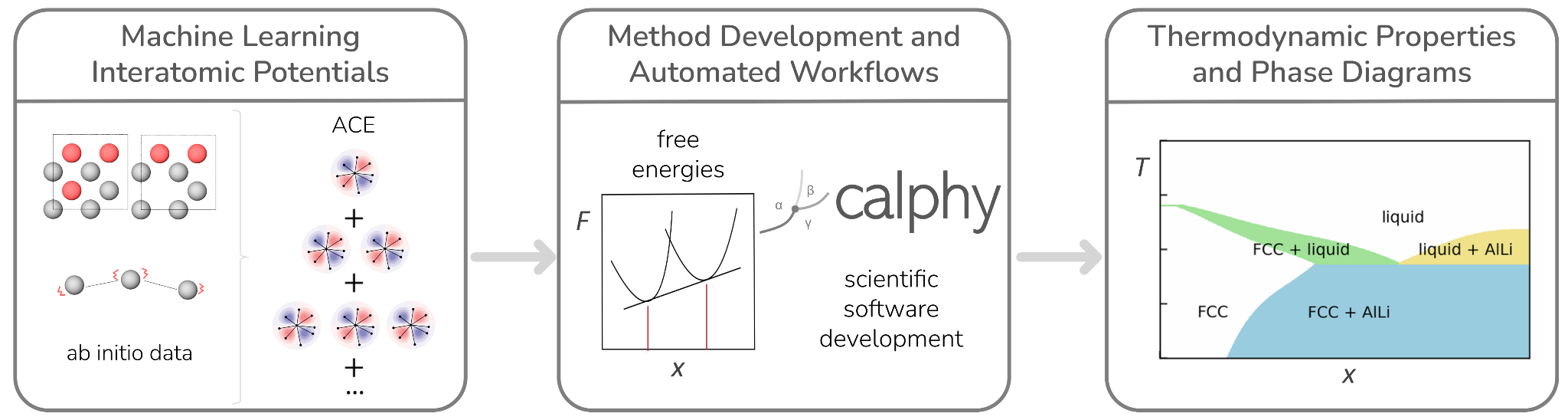
Our group develops computational methods for the accurate evaluation of thermodynamic quantities, with particular emphasis on free energy calculations for reliable phase diagram construction. We focus on machine learning interatomic potentials, in particular the atomic cluster expansion and its extensions (ACE, GRACE) to deliver ab initio accuracy at greatly reduced computational cost.
Another key activity of the group is the development and maintenance of scientific software and computational workflows that integrate every step of the research process in a reproducible and reusable manner.
Competences
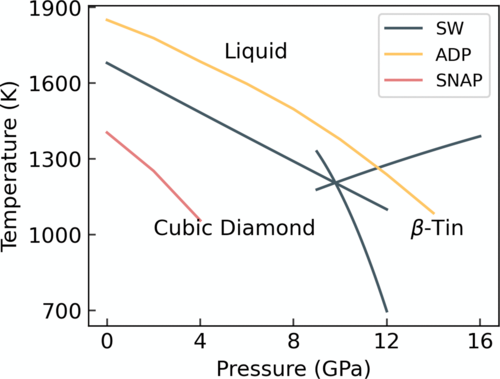
- Molecular dynamics calculations for phase diagrams
- Simulations of phase transitions
- Machine learning interatomic potentials and applications
- Reproducible computational workflows
- Scientific software development
Research Examples