Just another WordPress site - Ruhr-Universität Bochum
Research Group
Atomistic Simulation of Structural and Phase Stability
 RUB, Marquard
RUB, MarquardResearch Group Leader
Room: 02-571
Tel.: +49 234 32 29375
E-Mail: thomas.hammerschmidt@rub.de
Research
It requires adequate approaches to treat the diversity of the chemical compositions (e.g. multi-component superalloys), the complexity of the microstructures (e.g. dislocations and precipitates in steels), and the complexity of the physical phenomena (e.g. magnetic phase transition in iron, finite-T properties of battery materials, dislocations in high-entropy alloys).
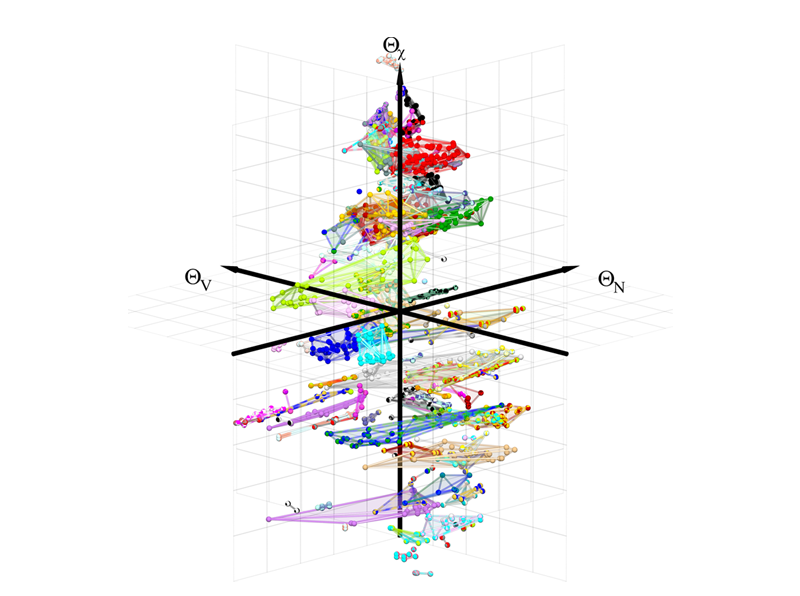
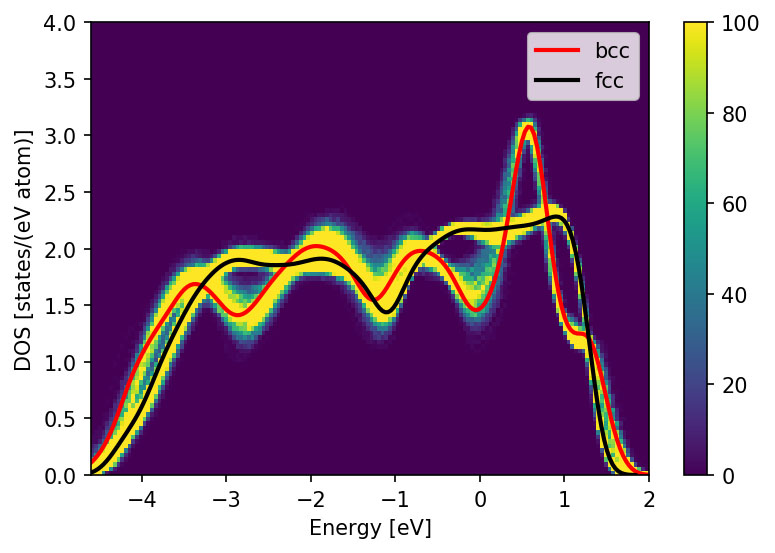
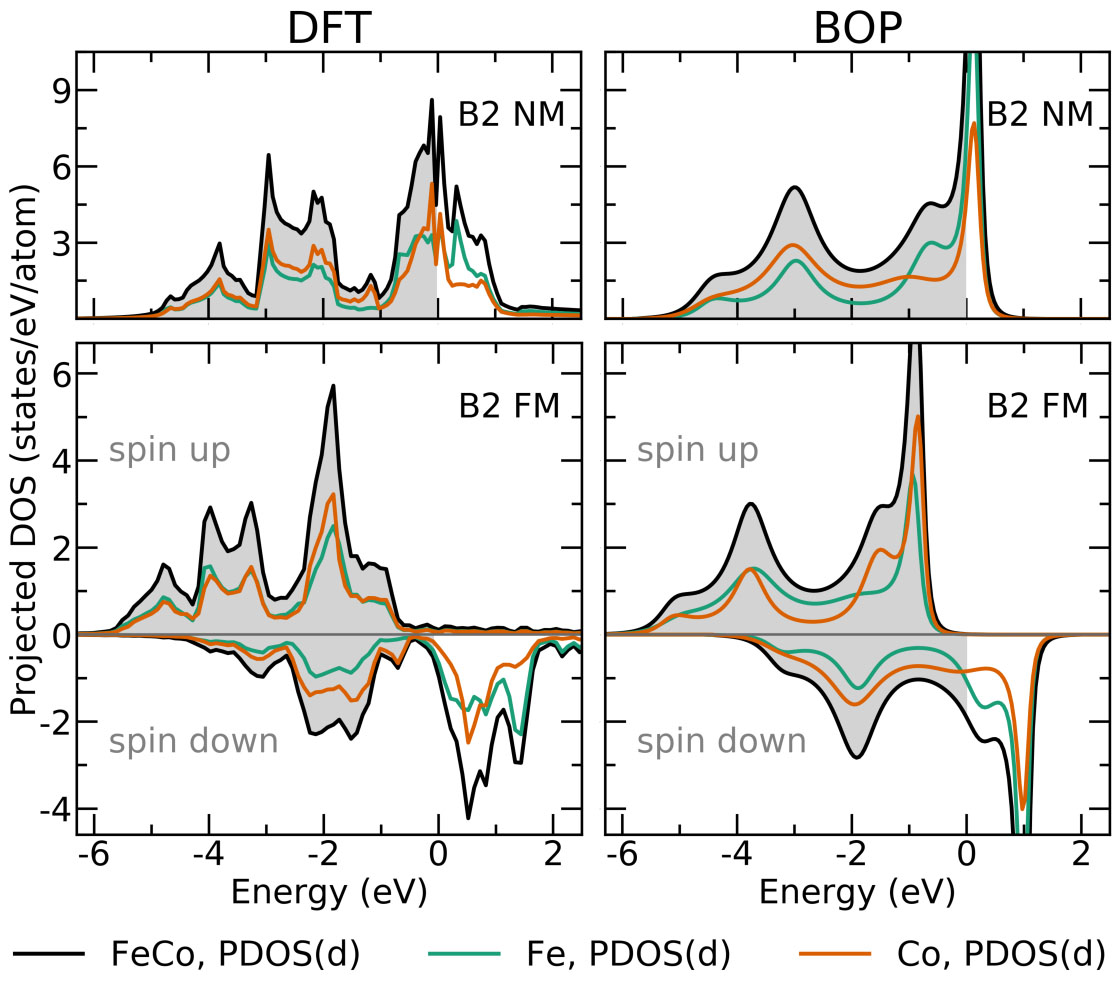
In our portfolio of materials-science methods, we combine electronic-structure methods at the level of density functional theory (DFT), tight-binding (TB), and analytic bond-order potentials (BOPs) with structure maps and machine-learning as data-driven methods. The TB/BOP models are obtained by coarse-graining the electronic structure, preserving the quantum-mechanical nature of the chemical bond for large-scale atomistic simulations that capture the complexity of microstructure and physical phenomena. They also provide electronic-structure-based descriptors of the local atomic environment, which are applied in the machine-learning of material properties across chemical space. The highly predictive structure maps chart the bonding chemistry of known compounds with physically intuitive descriptors and enable us to predict structural stability in multi-component alloys.
Competences
- Interatomic potentials based on physical models and machine learning
- Structure maps of d-d and p-d valent systems
- High-throughput density functional theory calculations
- Descriptors of local atomic environments and machine learning
- Structural stability, point defects and interfaces in transition metal compounds
Research Examples