Just another WordPress site - Ruhr-Universität Bochum
Research Group
Mechanical Properties of Interfaces
 RUB, Marquard
RUB, MarquardResearch Group Leader
Room: IC 02-503
Tel.: +49 234 32 29304
E-Mail: rebecca.janisch@rub.de
Research
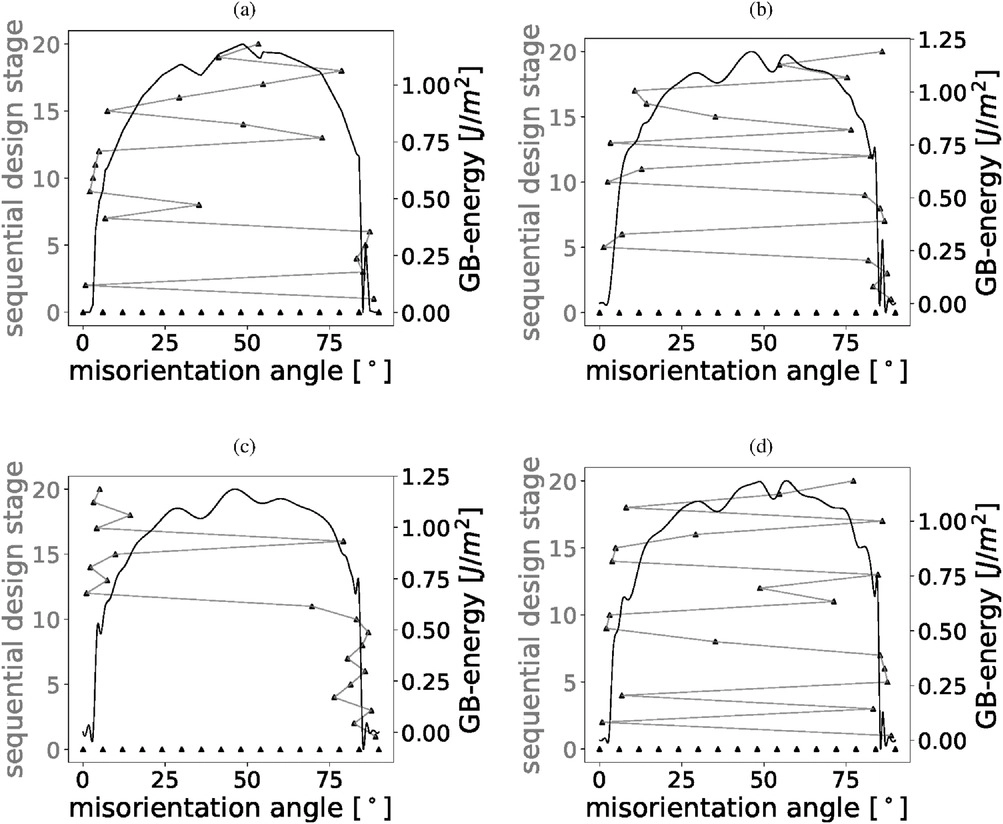
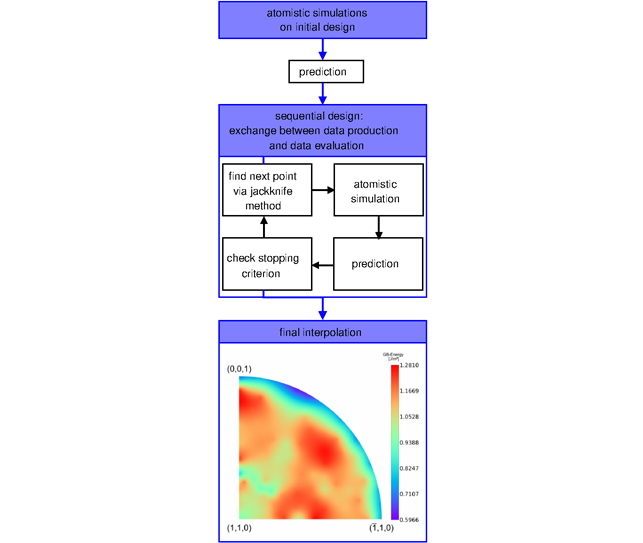
These processes are investigated by means of atomistic simulations. Ab-initio electronic structure calculations based on density functional theory are used to predict the energy, strength, and effective modulus of interfaces and other defects in iron and ferritic steel. Such characteristic properties are used to identify and understand trends, e.g. on hydrogen solubility in ferritic microstructures and on its effect on grain boundary cohesion. Thus, guidelines for alloy design and constitutive relationships for multiscale simulations, e.g. of hydrogen-enhanced decohesion, are derived. A combination of atomistic methods with a statistical assessment of the parameter space that defines the structure of grain boundaries allows a larger survey of structures in an efficient manner, which is needed to derive structure-property relationships.
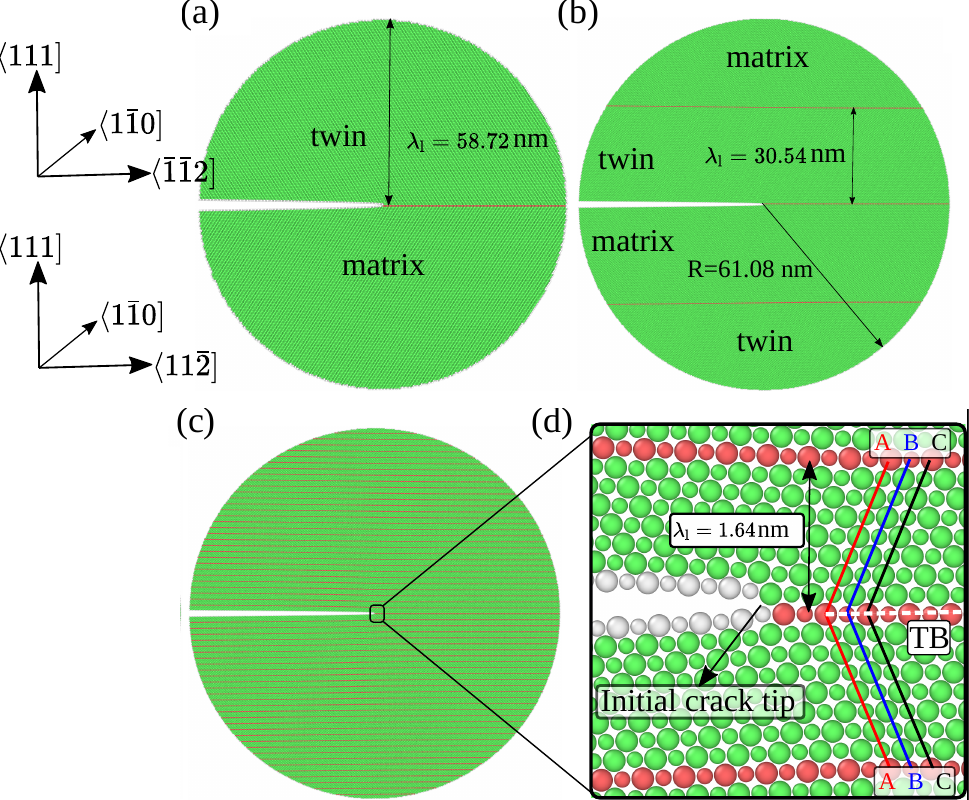
Via large-scale atomistic simulations the fundamental deformation and crack propagation mechanisms in interface-dominated microstructures are determined – such as interfacial sliding, migration, dislocation emission, and twinning in fully lamellar TiAl alloys – and are related to fundamental physical quantities, such as surface and stacking fault energies.
In the last years the group’s projects focused on the role of the excess volume of grain boundaries for the solution of H, the development of an efficient algorithm that combines design of experiment principles with atomistic simulations and the role of defects and crack geometry during fracture of lamellar and polycrystalline TiAl microstructures.
Competences
- Ab initio electronic structure calculations
- Molecular dynamics simulations
- Scale-bridging modelling of interface mechanics and thermodynamics
- Statistical methods
Research Examples