Just another WordPress site - Ruhr-Universität Bochum
Atomic Cluster Expansion (ACE) for carbon
Our extensive validation tests revealed that ACE is able to predict a broad range of properties of molecular, liquid, crystalline, and amorphous carbon phases while being several orders of magnitude more computationally efficient than available machine learning models. The outstanding predictive power of ACE was demonstrated on three distinct applications: a brittle crack propagation in diamond, an evolution of amorphous carbon structures at different densities and quench rates, and nucleation and growth of fullerene clusters under high pressure and temperature conditions.
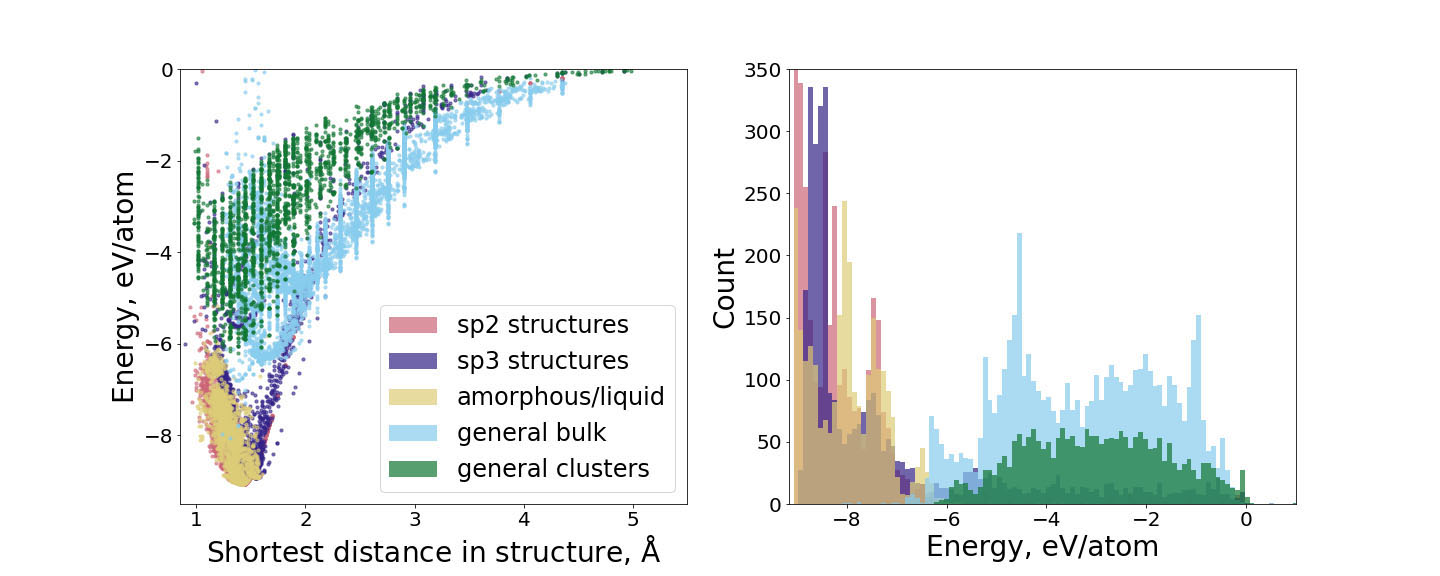
The key ingredient of constructing any ACE model is an accurate and consistent reference dataset that covers a large part of the phase space of atomic configurations. Such a dataset consists of a series of atomic structures and their corresponding energies, forces, and stresses, and is typically evaluated using electronic structure methods like DFT. In the case of carbon, several peculiarities exist related to its diverse chemical bonding. The visualization of the training data used for the construction of our carbon ACE parametrization is provided in Fig. 1. The dataset encompasses more than seventeen thousand structures that were chosen to sample a broad range of atomic configurations for carbon to ensure both accuracy and transferability.
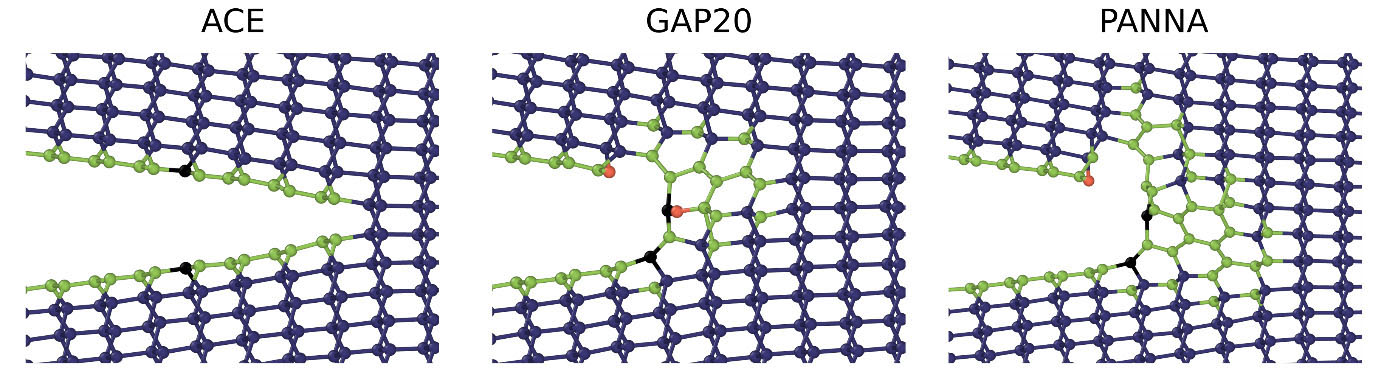
We simulated semi-infinite cracks with periodic boundary conditions applied along the crack front. The atomic configurations were generated by applying a given stress intensity factor KI to model the asymptotic crack tip region according to linear elastic fracture mechanics. Depending on the magnitude of the applied stress, the crack either tends to heal or to propagate during the simulation. Our simulations show that below the critical loading, all models predict the crack to close along the crack plane. However, ACE is the only model that sustains brittle cleavage when the loading exceeds the critical value of about 4.2 MPa m1/2, which is in good agreement with experimental observations. In contrast, both GAP20 and PANNA models show local structural transformations into graphitic structures, which lead to blunting the crack tip, as displayed in Fig. 2.
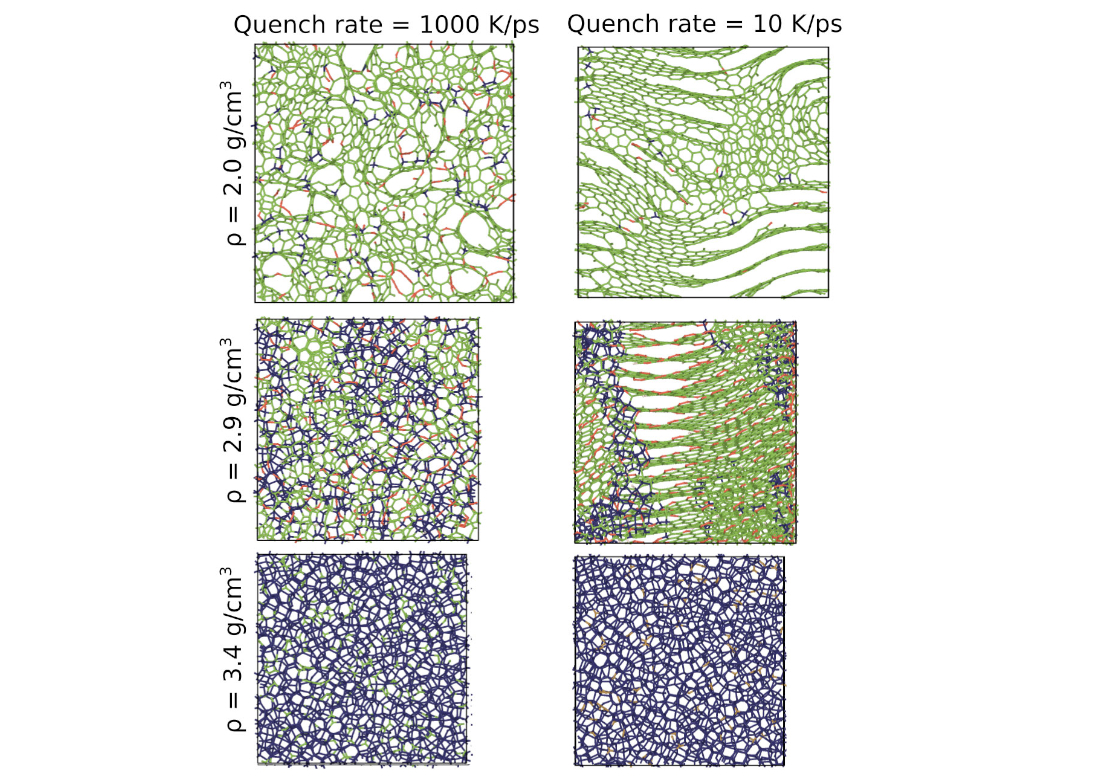
The great variability of amorphous carbon (a-C) networks, governed by competing sp, sp2 and sp3 hybridizations, poses another difficult challenge for atomistic simulations, and marked differences exist in the predictions of structural and physical properties of a-C systems from different atomistic models. We studied the properties of bulk a-C samples using large-scale molecular dynamics (MD) simulations. The samples with densities ranging from 1.8 (low-density nanoporous structures) to 3.5 (diamond-like a-C) g/cm3 were prepared using three different quench rates. Snapshots of representative structures are shown in Fig. 3.
Samples generated with the fast quench rate exhibit uniformly disordered structures that differ mainly in the fraction of the sp2 and sp3 bonded atoms. However, at the slow cooling rate, which was only possible to achieve due to the outstanding ACE efficiency, we observe an occurrence of more ordered structures with clearly separated sp2 and sp3 regions. For the lower densities, slow quenching leads to extended graphitic sheets with fewer defects. Hence, our simulations demonstrate for the first time a clear influence of the quench rate on the a-C morphology with a non-classical potential.
In summary, our carbon ACE opens new possibilities for structural modelling of carbon at the atomic scale. It not only describes the fundamental properties of carbon allotropes with DFT accuracy but is also able to maintain this accuracy in large-scale simulations. If necessary, the ACE accuracy and transferability can be further improved systematically, either by tailoring the training dataset for the required application or by extending the ACE basis. Finally, the elemental ACE models can be readily extended or combined to address multicomponent systems, such as hydrocarbon systems or transition metal carbides.