Just another WordPress site - Ruhr-Universität Bochum
Research Group
Atomistic Simulation of Mechanical Behaviour
 RUB, Marquard
RUB, MarquardResearch Group Leader
Room: IC 02-571
Tel.: +49 234 32 29313
E-Mail: matous.mrovec@icams.rub.de
Research
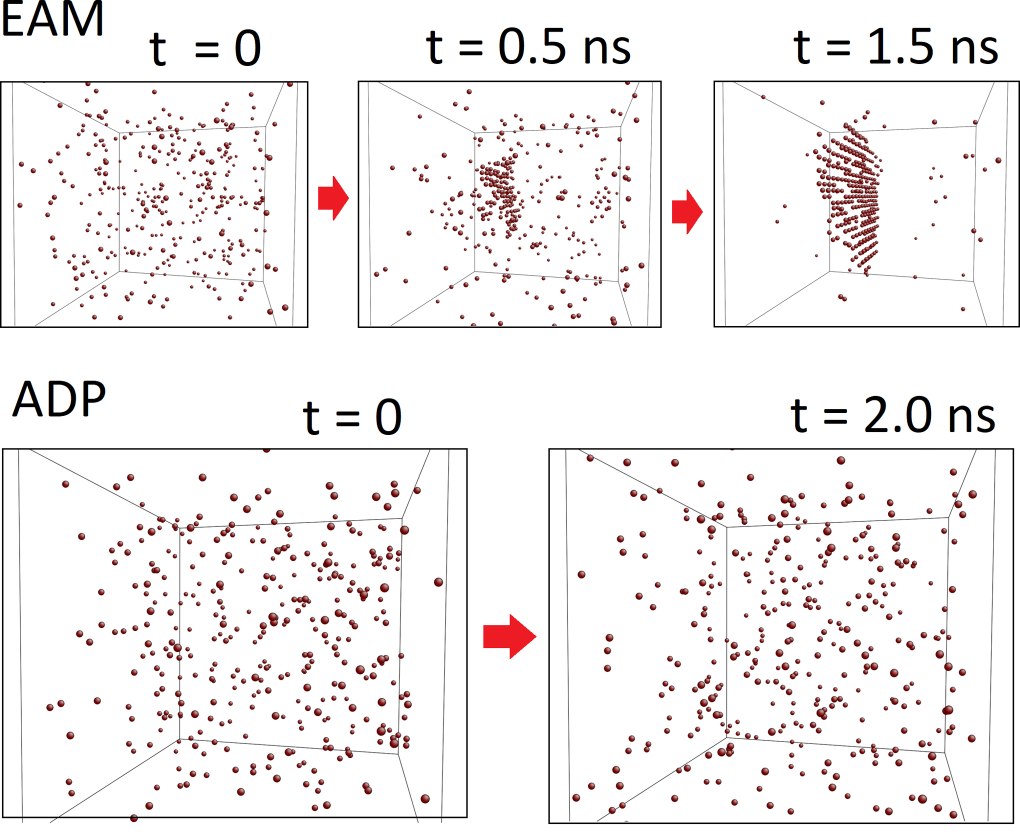
We start with modelling intrinsic material properties related to chemical bonding but eventually concentrate on the role of crystal imperfections. The imperfections encompass fundamental crystal defects such as vacancies, dislocations, and grain boundaries in single-component crystalline materials as well as complex microstructural features such as semicoherent interfaces, precipitates, and secondary phases that constitute the microstructure of technologically important multi-phase and multi-component systems.
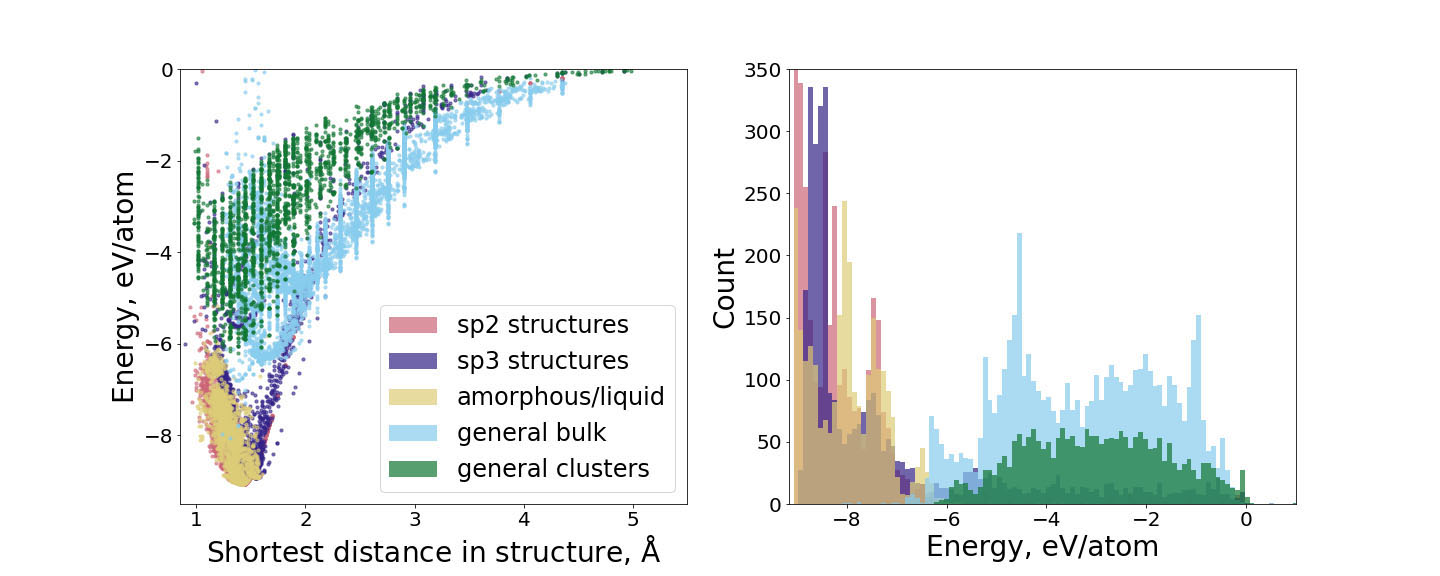
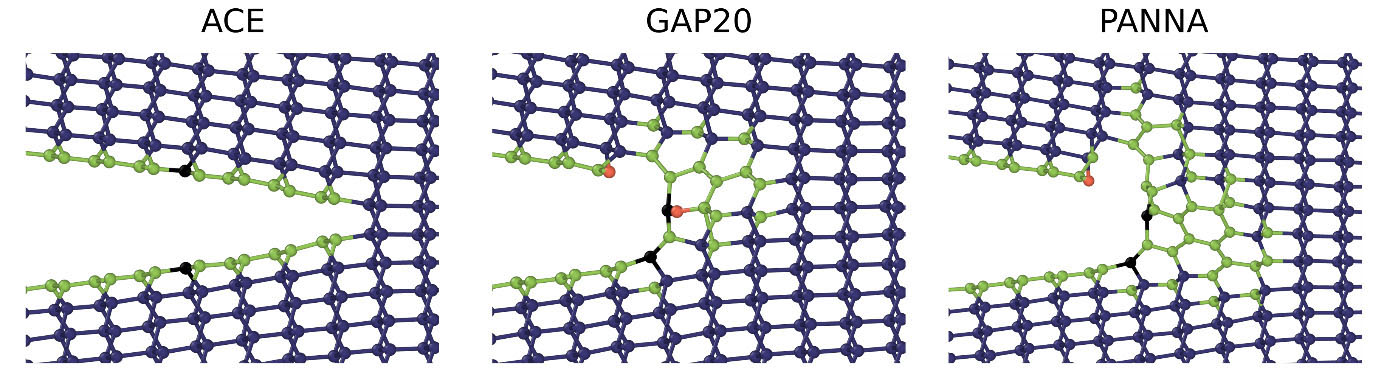
We are interested in materials with prototypical metallic and covalent chemical bonding as well as in those with mixed metallic-covalent or covalent-ionic character such as transition metals and their compounds, perovskite oxides, and carbon-based materials. The methods and models we employ span the whole atomistic modelling hierarchy from accurate first-principles methods through approximate electronic structure approaches to novel interatomic potentials. Recently, we have focused on the development and application of atomic cluster expansion (ACE) models that can reach the accuracy and transferability of electronic structure methods while remaining highly computationally efficient and applicable in large-scale atomistic simulations. We also integrate atomistic simulations and mesoscale techniques (DDD, kMC), phenomenological and continuum theories as well as experiments.
Competences
- Interatomic potentials
- Transition metals and their compounds
- Crystal defects and imperfections
- Hydrogen embrittlement
- Magnetism
Research Examples